История
Клоакальная экстрофия впервые описана Littre в 1709 году, однако первая успешная реконструкция произведена лишь в 1960 году Rickham. До этого все больные с данным пороком, тяжелым и сложным, были неизбежно обречены на гибель, ибо слишком большие затраты на лечение, эмоциональные, физические и экономические, удерживали хирургов от попыток осуществления сложных реконструктивных операций.
Первые операции при клоакальной экстрофии сопровождались очень высокой летальностью. По данным сводной статистики, среди 19 больных, оперированных до 1970 года, выжили только 6. В Бостонском Детском Госпитале из 34 пациентов, лечившихся с 1968 по 1976 год, выжили лишь 17. С общим прогрессом в неонатальной интенсивной терапии и в разработке проблем питания появились и более обнадеживающие сообщения о лечении клоакальной экстрофии. В Детском Госпитале Филадельфии из 15 больных выжили 13.
Согласно еще одной публикации, выживаемость при этом пороке увеличилась с 22% в 1963—1978 гг. до 90% в 1979—1986 гг. Примерно такие же результаты сообщаются и другими авторами, у которых за 22 года лечились 12 детей и выживаемость составила 83%. Rickham, несомненно, был проницателен, предсказывая, что «. в следующем десятилетии детской хирургии придется вплотную столкнуться с данной проблемой. И мы вынуждены будем пересмотреть устоявшееся представление о том, что эти дети никогда не могут стать полноценными членами общества, а потому им лучше умереть».
Анатомия
Клоакальная экстрофия, обозначаемая также названиями: пузырно-кишечная расщелина, эктопическая клоака, висцеральная эктопия, осложненная экстрофия мочевого пузыря, расщелина брюшной стенки, — является наиболее тяжелой формой аномалии брюшной стенки. Комплекс анатомических изменений при классическом варианте клоакальной экстрофии показан на рисунках. Он включает в себя грыжу пупочного канатика наверху, открывающиеся наружу кишечник и мочевой пузырь — внизу.
Мочевой пузырь расщеплен на две части по средней линии участком кишечной слизистой, причем каждый «полупузырь» имеет выходное отверстие мочеточника. Кишечная слизистая, расположенная между половинами мочевого пузыря, гистологически представляет собой илеоцекальную область и может иметь несколько отверстий (до четырех).
Самое верхнее отверстие относится к проксимальному отделу кишечника и может пролабировать в виде «хобота слона», в то время как наиболее низко расположенное отверстие ведет в дистальную часть кишечника. Дистальная кишка представляет собой каудальный отдел (задняя кишка), заканчивающийся слепо и сочетающийся с атрезией ануса. Между проксимальным и дистальным отверстиями могут располагаться еще одно или два «аппендикулярных» отверстия.
Во всех случаях имеются также аномалии половых органов. У мальчиков это неопущение яичек, расщепленный половой член, каждая половина которого имеет еще и эписпадию и тесно соединена с широко «расставленными» лонными областями. У девочек обычно расщеплен клитор, удвоено влагалище и имеется двурогая матка.
Эмбриогенез
Для того, чтобы понять, как возникает клоакальная экстрофия, необходимо знать нормальную эмбриологию клоакальной области. Поскольку человеческий эмбрион не проходит через стадию развития, соответствующую экстрофии, то существует вполне логичное предположение о том, что клоакальная экстрофия возникает в результате именно нарушений эмбрионального развития, а не как следствие просто задержки (остановки) нормальных процессов в эмбриогенезе.
Клоакальная мембрана, отделяющая на ранних стадиях развития целомическую полость от амниотического пространства, появляется между 2-й и 3-й неделями гестации. В это время она определяется по средней линии непосредственно каудальнее примитивной полоски в виде участка, где эктодерма соединяется с эндодермой без посредничества мезодермы. К 4-й неделе, по мере удлинения эмбриона и его хвоста, клоакальная мембрана образует вентральную стенку урогенитального синуса в основании аллантоиса. Неслившиеся между собой закладки половых бугорков лежат краниально и латерально по отношению к клоакальной мембране.
В конце концов эти закладки увеличиваются и сливаются по средней линии выше клоакальной мембраны, образуя половой бугорок. Это слияние сопровождается врастанием (по направлению к средней линии) мезодермы, в результате чего удлиняется участок между целомом и клоакальной мембраной и появляется возможность для развития интактной инфраумбиликальной брюшной стенки. В это же время уроректальная перегородка развивается в каудальном и медиальном направлениях, соединяясь с примитивной промежностью (клоакальная мембрана) и разделяя клоаку на урогенитальный синус и прямую кишку.
Две основные теории объясняют аномальный эмбриогенез клоакальной экстрофии. Согласно первой из них — теории «каудального смещения» — к возникновению спектра аномалий, составляющих комплекс «эписпадия—экстрофия», приводит каудальное смещение парных закладок половых бугорков относительно того участка, где уроректальная складка делит примитивную клоаку на урогенитальный и анальный компоненты. Слияние закладок по средней линии в месте, где урогенитальная перегородка соединяется с клоакальной мембраной, приводит к эписпадии.
Продолжающееся каудальное смещение закладок может быть причиной возникновения полной экстрофии мочевого пузыря, а еще более дистальное (и равномерное) их смещение (каудальнее по отношению к анальной части клоаки) приводит к экстрофии и кишечника, и мочевого пузыря и таким образом — к клоакальной экстрофии. Степень каудального смещения парных закладок половых бугорков может быть различной, и в зависимости от этого встречается разный спектр пороков.
Другая ведущая теория объясняет развитие клоакальной экстрофии «эффектом клина», создаваемого аномально большой клоакальной мембраной. Приверженцы этой теории считают, что если бы генез данного порока был связан лишь с простым каудальным смещением парных закладок, то в результате возникала бы скорее и чаще всего нетяжелая степень эписпадии. Однако почти у 90% пациентов с клоакальной экстрофией аномалии локализуются в области выходного отдела мочевого пузыря. Кроме того, при дальнейшем каудальном смещении закладок и более тяжелой экстрофии пещеристые тела должны бы были локализоваться на промежности, не будучи соединенными с лонной костью. Однако подобный вариант порока описывается редко.
Согласно данной теории, аномально большая или персистирующая клоакальная мембрана играет роль клина для развивающихся структур брюшной стенки. Это предположение вполне логично объясняет, почему вовлеченные в аномалию анатомические структуры нормально развиты, но расщеплены на две части, и почему неправильно развиты кости — это может быть связано с различной степенью эффекта клина. Если разрыв непрочной мембраны произошел до опускания уроректальной перегородки и слияния половых бугорков, то образуется инфраумбиликальный дефект по средней линии, обнажающий слизистую мочевого пузыря и кишечника и обусловливающий расщепление наружных гениталий.
Правомерность теории эффекта клина подтверждается экспериментально. Инертные пластические трансплантаты помещались в область клоакальной мембраны у развивающегося куриного эмбриона. Трансплантат обеспечивал эффект клина а также персистирование клоакальной мембраны, и в результате вызывал развитие инфраумбиликальных дефектов различной степени выраженности. Многие полученные в эксперименте дефекты по размерам были намного больше самого трансплантата. Это первая созданная модель экстрофии представляла собой аномалию, которая в норме не встречается у цыплят.
Хотя данные теории и объясняют процесс развития срединных дефектов и роль при этом аномальной клоакальной мембраны, однако остается, к сожалению, не ясно, каковы же причины возникновения илеоцекального пролапса и атрезии ануса и почему илеоцекальный отдел кишечника слепо заканчивается. По одной из теорий, указанные виды пороков обусловлены резким замедлением (в результате вовлечения в экстрофию) роста задней кишки, начинающейся на уровне дивертикула Меккеля. Открывающаяся наружу кишка представляет собой чрезвычайно короткую заднюю кишку, а дистальный, слепо заканчивающийся отдел — персистирующую хвостовую (постанальную или постклоакальную) кишку.
Альтернативное объяснение, согласно другой теории, состоит в том, что петля средней или задней кишки пролабирует между половинами мочевого пузыря и оказывается странгуляционно сдавленной. Данное предположение вполне достоверно объясняет причину укорочения кишечника, а также наличие множественных отверстий на его поверхности.
Чтобы объяснить отсутствие у некоторых пациентов с клоакальной экстрофией дериватов дисталь-ного сегмента средней кишки (терминальный отдел ileum, аппендикс, слепая кишка и правая половина colon), некоторые исследователи высказывают предположение о том, что определенную роль в развитии дистальных отделов средней кишки играет эпителий аллантоисного происхождения. Задержка краниальной миграции этого эпителия и его соединения с желточным мешком или персистенция эпителия в области свода клоаки может быть причиной возникновения данного типа экстрофии.
Поскольку развитие клоакальной экстрофии — очень сложный процесс, то не каждый случай этого порока анатомически представляет собой классический его вариант. Предложена классификация экстрофии, которая позволяет более правильно и логично оценивать и анализировать различные ее формы.
Закрытая клоакальная экстрофия обладает всеми чертами висцеральных и мышечно-скелетных аномалий, присущих клоакальной экстрофии, но при этом варианте брюшная стенка интактна. В нашей практике встретились двое больных с «закрытой клоакальной экстрофией». Внешне порок выглядел как закрытая экстрофия мочевого пузыря, а на операции обнаружено, что мочевой пузырь сзади представлен илеоцекальной частью кишечника. В этих случаях была также короткая задняя кишка и атрезия ануса.
Эмбриологически отсутствие миграции мезодермы в инфраумбиликальную мембрану приводит к разрыву последней, в результате чего органы малого таза оказываются открытыми на поверхности живота. Закрытая клоакальная экстрофия возникает в тех случаях, когда мезодерма внедряется в инфраумбиликальную мембрану, предупреждая ее разрыв, но уже после того, как клиновидный эффект мембраны привел к расщеплению лонных костей и латеральной мускулатуры брюшной стенки. Эта мезодермальная инвазия может быть неполной или полной, а по времени либо предшествовать выворачиванию наружу внутренних органов либо возникать вслед за ним.
Наличие абдоминальных мышечно-скелетных дефектов при нормальном развитии внутренних органов объясняется поздним полным мезодермальным внедрением до выворачивания внутренних органов. Позднее, но неполное внедрение мезодермы может вести к формированию таких вариантов пороков, как верхняя пузырная расщелина, когда экстрофирована только верхушка мочевого пузыря или когда кишечник и мочевой пузырь экстрофированы, но в пределах субумбиликальной области, а уретра и половой член при этом нормально развиты.
При экстрофии или эписпадии как у мальчиков, так и у девочек, может отмечаться секвестрация сегментов внутренних органов (толстой или подвздошной кишки) на брюшную стенку, что объясняется внутриутробной задержкой мезодермальной инвазии со вторичным закрытием (полным или неполным) разорвавшейся ранее инфраумбиликальной мембраны. Секвестрированные сегменты кишки могут быть использованы при хирургической реконструкции мочеполового тракта.
Частота
Клоакальная экстрофия представляет собой крайнюю степень спектра аномалий, включающего в себя самые разнообразные пороки — от эписпадии до клоакальной экстрофии. На эписпадию разных видов, встречающуюся с частотой 1:120 000 рождений, приходится около 30% пороков этой группы. Классическая экстрофия мочевого пузыря является наиболее частой (60%) среди этих аномалий — 1:30 000 рождений. Остальные 10 % падают на клоакальную экстрофию (и ее варианты), которая отмечается у одного на 400 000 новорожденных. В Соединенных Штатах ежегодно можно ожидать рождения 15 детей с этим пороком. Аномалии данной группы отчетливо преобладают у мальчиков. Особенно это касается классической экстрофии мочевого пузыря, где соотношение мальчики — девочки составляет 2,3:1. И меньше всего половые различия отмечаются в группе клоакальной экстрофии.
Даже фантасты подчас не способны придумать то, что способна сотворить природа.
При первых признаках простуды считаете себя самым больным человеком в мире и требуете повышенного к себе внимания? Вы просто не знаете, с какими заболеваниями живут некоторые люди! Не зря говорят, все познается в сравнении, сообщает Day.Az со ссылкой на kp.ru.
В двух словах, это много-много бородавок по всему телу. Такое состояние возникает, когда человек имеет чрезвычайную чувствительность к ВПЧ (вирус папилломы человека) и это считается генетическим заболеванием. Самое неприятное в таком состояние даже не столько то, как человек выглядит с таким недугом, сколько повышенный риск развития рака кожи, потому что ультрафиолетовый свет ухудшает состояние больного. Такие люди обречены постоянно носить защитную одежду и использовать солнцезащитный крем.
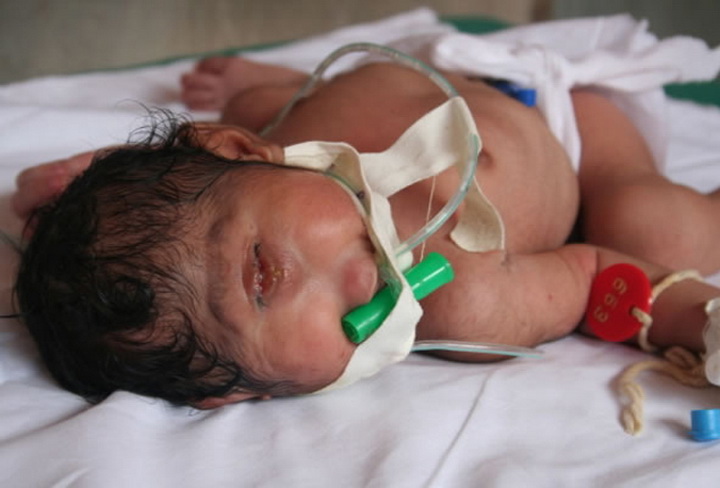
То есть один глаз вместо двух. Этот порок возникает, когда во время формирования еще в утробе матери глазные яблоки сращиваются и помещаются в одной глазнице. Такое заболевание считается несовместимым с жизнью, циклопы погибают в первые же дни после рождения.
Или, чтобы было понятнее, синдром русалки. Так называется аномалия, при которой человек рождается со сращенными нижними конечностями. Из-за этого у людей часто отсутствуют наружные половые органы и недоразвит желудочно-кишечный тракт и анус. На фото ниже изображена Шило Пепин, она родилась с диагнозом "сиреномелия", и врачи давали ей шансы на жизнь всего в всего несколько дней. А она прожила десять лет. Умерла 2008 году.
Чрезмерный рост волос, которые покрывают зачастую все тело. В том числе на участках кожи, где их быть не может вообще. Считается заболеванием преимущественно женщин. Причиной может стать заболевание нервной системы, например, опухоль головного мозга, или тяжелый стресс, некоторые инфекционные болезни, нарушение обмена веществ. А также нарушения гормонального фона, некоторых желез (яичники, надпочечники, гипофиз), проблемы с щитовидкой.
Так называется явление, когда кожа людей становится голубого цвета. Почему они такие? Из-за того, что в крови повышается количество метгемоглобина. Собственно, отсюда и название диагноза. Метгемоглобин плохо переносит кислород от легких к тканям организма, в результате развивается тканевая гипоксия, а кожа приобретает синий оттенок. Может развиться также в результате некоторых видов острых химических отравлений.

СИНДРОМ ЮНЕР ТАНА
Люди, способные передвигаться только на четырех конечностях, а не "на своих двоих". У них также наблюдается выраженная задержка умственного развития — сканирование мозга у таких людей показало, что он имеет упрощённую структуру, отсутствуют целые участки, которые есть у остальных людей. Ученые сходятся во мнении, что такие явления возникают в результате генетического сбоя.

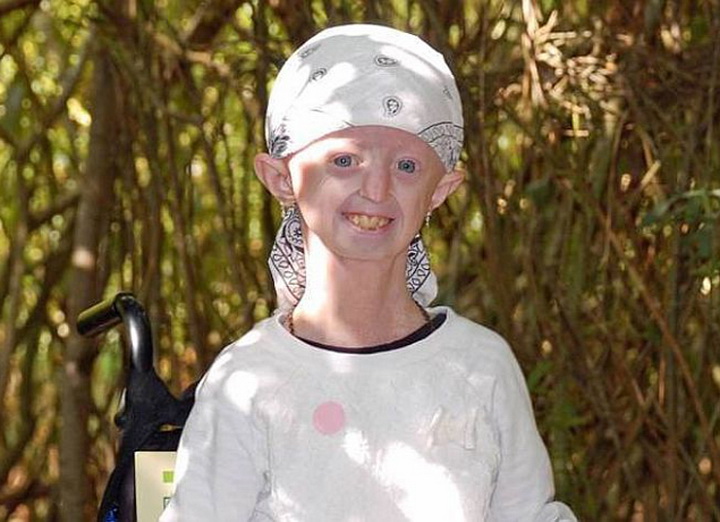
Очень редкий случай. Например, последний связан с Хэйли Окинс, 17-летней девочкой с телом 104-летней старушки из Великобритании. Она старилась в восемь раз быстрее обычных людей. По словам мамы девочки Керри Окинс, врачи пообещали, что малышка, в лучшем случае, доживет до 13 лет. Но Хэйли оказалась настоящим бойцом. Она и ее семья всячески стремились обратить внимание общественности на проблему детей с прогерией. Хэйли много путешествовала и встречалась со знаменитостями, среди которых Кайли Миноуг и Принц Чарльз. И вела страничку в Facebook. А в апреле этого года умерла.
Протей, герой древнегреческой мифологии, мог менять форму своего тела. Однако люди с диагнозом его имени, к сожалению, сами свои тела не меняют, их тела меняются сами. Джозеф Меррик, живший в Лондоне в 19-ом веке и получивший прозвище "человек-слон", тоже из числе людей с синдромом Протея.
В своей "Автобиографии" он так описал себя: ". часть моей головы покрыта, так сказать, холмами и долинами, и это всё смешано в общую кучу, в то время как лицо имеет такой вид, что никто не мог описать его. Правая рука — имеет размер и форму слоновьего хобота, другая рука — не больше чем рука девочки десяти лет, хотя она и вполне работоспособная. Мои ноги и ступни охвачены толстой шероховатой кожей, подобно коже слона, и имеет почти тот же самый цвет. Никто бы не поверил, что такое может существовать, пока не увидел это".

Заболевание, при котором от человека исходит неприятный запах, напоминающий запах гниющей рыбы. Иногда явление так и называют — синдром рыбного запаха. Отчего такие "ароматы"? Ничего общего в редкими походами в душ. Просто в организме таких людей накапливается много триметиламина, он и издает такой неприятный запах. В принципе, это не вредно, но психологически оч-ч-чень неприятно.

Редкое генетическое заболевание, при котором организм начинает формировать новые кости, да еще в неположенных местах — внутри мышц, связок, сухожилий и других соединительных тканей. К их образованию может привести любая травма: ушиб, порез, перелом, внутримышечная инъекция или операция. А удалять новые косточки нельзя, иначе могут только сильнее разрастись. У болезни нет расовой, половой, географической предрасположенности. Чаще всего она возникает из-за мутации.
На ногах у таких людей не по 5 пальцев, как у всех остальных, а всего лишь по два. И оба являются большими. Ученые предполагают, что такое необычное строение ступни было вызвано каким-то неизвестным вирусом. Либо же вызвано интимными кровными связями в рамках одного семейства.











—> —>
Заметили ошибку в тексте? Выберите текст и сообщите нам, нажав Ctrl + Enter на клавиатуре
Испокон веков людей, имеющих такие мутации, клеймили как уродов и монстров. Однако сегодня мы знаем, что необычный внешний вид — лишь часть широкого спектра генетических вариаций нашего вида. Здесь мы осветим 10 самых интересных, на мой взгляд, случаев


Большинство детей, больных прогерией, умирают в возрасте около 13-ти лет, но некоторые доживают и до 20-ти. Как правило, причиной смерти становится сердечный приступ или инсульт. В среднем, прогерия случается только у одного ребёнка из 8 000 000.Заболевание вызвано мутациями в гене ламин A/C, белке, обеспечивающем поддержку клеточным ядрам. Другие симптомы прогерии включают жёсткую кожу, полностью лишённую волосяного покрова, костные аномалии, замедление роста и характерную форму носа. Прогерия представляет большой интерес для геронтологов, которые надеются выявить связь между генетическими факторами и процессом старения
Синдром Юнера Тана

Синдром Юнера Тана (СЮТ) характерен прежде всего тем, что люди, страдающие им, ходят на четвереньках. Открыл его турецкий биолог Юнер Тан после изучения пяти членов семьи Улас в сельской местности Турции. Чаще всего люди с СЮТ пользуются примитивной речью и имеют врождённую мозговую недостаточность. В 2006-м году о семье Улас был снят документальный фильм под названием «Семья, ходящая на четвереньках». Тан описывает это так:«Генетическая природа синдрома предполагает обратную ступень в эволюции человека, вызванную, скорее всего, генетической мутацией, обратному процессу перехода от квадропедализма (хождения на четырёх конечностях) к бипедализму (хождению на двух). В этом случае синдром соответствует теории прерывистого равновесия.Новый синдром, по словам Тана, может быть использован в качестве живой модели человеческой эволюции. Некоторые исследователи, впрочем, не относятся к этому серьёзно и считают, что проявление СЮТ зависит не от генома

Гипертрихоз также называют «синдромом оборотня» или «синдромом Абрамса». Он проявляется только у одного человека из миллиарда, и только 50 случаев со времён Средневековья были задокументированы. Люди, страдающие гипертрихозом, отличаются чрезмерным количеством волос на лице, ушах и плечах. Это происходит из-за нарушения связей между эпидермисом и дермой во время формирования у трёхмесячного плода волосяных фолликул.Как правило, сигналы от образующейся дермы «сообщают» фолликулам их форму. Фолликулы тоже, в свою очередь, сигнализируют кожным слоям, что в этой области одна фолликула уже есть, и это приводит к тому, что на теле волоски растут на приблизительно одинаковом расстоянии друг от друга. В случае с гипертрихозом эти связи нарушены, что приводит к образованию слишком плотного волосяного покрова на тех участках тела, где его быть не должно

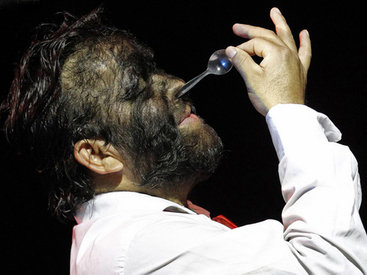
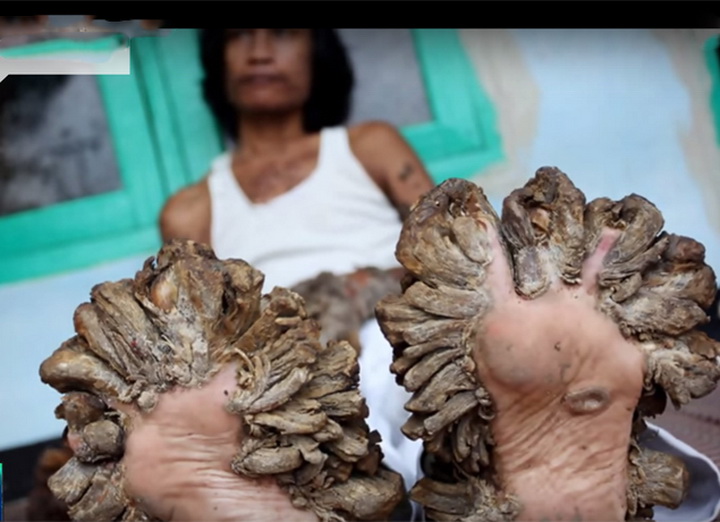
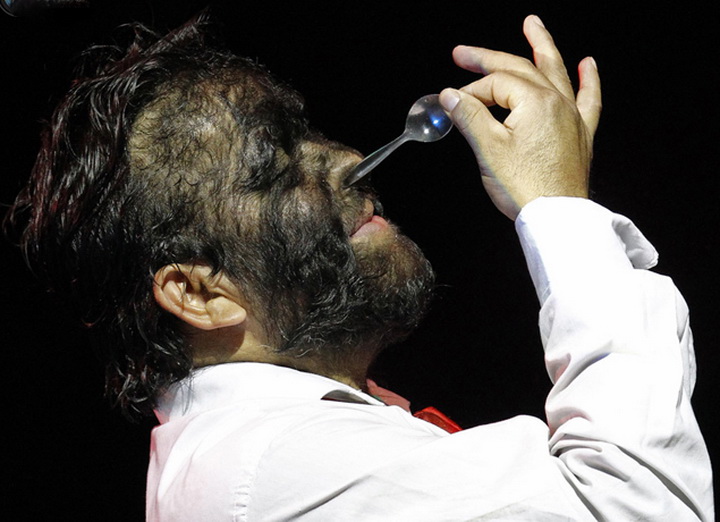
Эпидермодисплазия верруциформная — отклонение чрезвычайно редкое, делающее своих носителей склонными к широко распространённому вирусу папилломы человека (ВПЧ). Эта инфекция вызывает образование на коже чешуйчатых пятен и папул (плоскоклеточный рак кожи), растущих на руках, ногах и даже лице. Эти «наросты» выглядят как бородавки или чаще напоминают рог или древесину. Как правило, опухоли на коже начинают проявляться у людей в возрасте от 20 до 40 лет в местах, открытых для солнечных лучей. Методов полного исцеления не существует, однако с помощью интенсивной терапии можно уменьшить или на некоторое время приостановить распространение наростов.Общественность узнала об этом генетическом заболевании в 2007-м году, когда в интернете появилось видео с 34-летним индонезийцем Деде Косварой. В 2008-м мужчина перенёс операцию по удалению шести кг наростов с тела. Роговые образования были сняты с рук, головы, туловища и ног, и в эти места была пересажена новая кожа. В общей сложности удалось избавить Косвара от 95% бородавок. К сожалению, через некоторое время они снова начали расти, и врачи полагают, что операцию придётся повторять каждые два года, чтобы Косвара хотя бы мог держать ложку
Тяжёлый комбинированный иммунодефицит

Люди с этим генетическим отклонением рождаются без эффективной иммунной системы. Болезнь стала известна после вышедшего на экраны в 1976-м году фильма «Мальчик в пластиковом пузыре», вдохновлённого жизнью двух мальчиков-инвалидов Дэвида Веттера и Теда ДеВиты. Главный герой, маленький мальчик, вынужден жить в изолированной от окружающего мира пластиковой кабинке, поскольку нефильтрованный воздух и воздействие микроорганизмов могут оказаться для него смертельными. Реальный Веттер смог прожить таким образом до 13-ти лет, но умер в 1984-м году после неудачной трансплантации костного мозга — врачебной попытки укрепить иммунитет.Расстройство вызвано целым рядом генов, включая те, которые вызывают дефекты в Т и Б клеточных откликах, что в итоге оказывает негативное влияние на выработку лимфоцитов. Считается также, что это заболевание возникает в связи с отсутствием аденозиндезаминазы. Сейчас известны некоторые методы лечения с помощью генной терапии

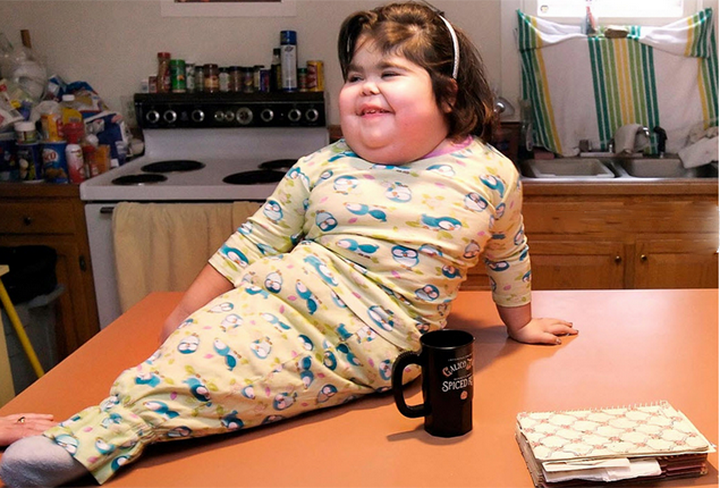
СЛН проявляется у одного младенца мужского пола из 380 000 и приводит к увеличению синтеза мочевой кислоты. Мочевая кислота выделяется в кровь и мочу в результате происходящих в организме химических процессов. У людей с СЛН в кровь поступает слишком много мочевой кислоты, которая накапливается под кожей и в итоге вызывает подагрический артрит. Кроме того, это может привести к образованию камней в почках и мочевом пузыре.Заболевание также влияет на неврологические функции и поведение. У страдающих СЛН часто непроизвольно сокращаются мышцы, что выражается как судороги и/или беспорядочное размахивание конечностями. Бывает, что больные калечат сами себя: бьются головой о твёрдые предметы, кусают пальцы и губы. От подагры может помочь аллопуринол, но методов лечения неврологических и поведенческих аспектов заболевания не существует

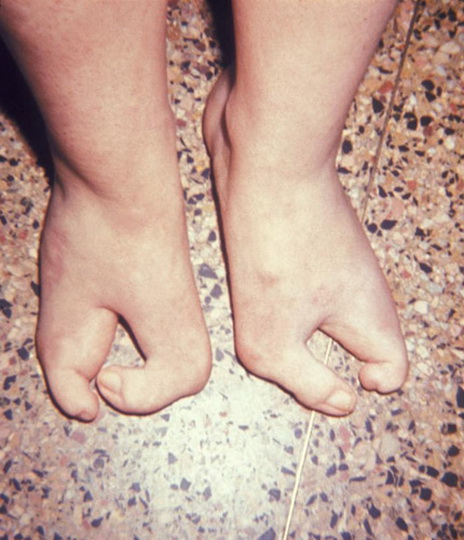
У страдающий эктродактилией пальцы на ногах или руках либо отсутствуют, либо недоразвиты, из-за чего кисти рук или ступни напоминают клешни. К счастью, такие нарушения в геноме встречаются редко. Эктродактилия может проявляться по-разному, иногда пальцы просто срастаются, и в этом случае их можно разделить с помощью пластической операции, в других случаях пальцы даже не сформированы до конца. Часто заболевание сопровождается полной потерей слуха. Причины болезни — нарушения генома, в том числе делеции, транслокации и инверсии в седьмой хромосоме

Вероятно, именно от этого заболевания страдал Джозеф Меррик, известный как Человек-слон. Синдром Протея вызван нейрофиброматозом типа I. При синдроме Протея кости и кожный покров больного могут начать увеличиваться аномально быстро, в результате чего нарушаются естественные пропорции тела. Обычно признаки заболевания не проявляются раньше 6–18 месяцев после рождения. Тяжесть заболевания зависит от индивидуума. В среднем синдромом Протея страдает один человек из миллиона. За всю историю задокументировано всего несколько сотен подобных случаев.Расстройство — результат мутации в гене AKT1, ответственном за регуляцию клеточного роста, в результате чего некоторые мутировавшие клетки растут и делятся с невообразимой скоростью, а другие клетки продолжают расти в нормальном темпе. В итоге получается смесь нормальных и ненормальных клеток, что вызывает внешние аномалии

Это генетическое заболевание встречается настолько редко, что уровень заболеваемости даже не известен. Но если кто-то находящийся рядом с вами страдает этим — вы сразу заметите. Дело в том, что в организме больного накапливается триметиламин, который, выделяясь вместе с потом, создаёт неприятный запах — от человека пахнет тухлой рыбой, тухлыми яйцами, мусором или мочой.Женщины, как правило, подвержены заболеванию в большей степени, чем мужчины. Интенсивность запаха достигает своего пика непосредственно перед и во время менструации, или после приёма оральных контрацептивов. Судя по всему, это связано с женскими половыми гормонами вроде прогестерона и эстрогена. Разумеется, в результате больные часто подвержены депрессии и предпочитают жить в изоляции

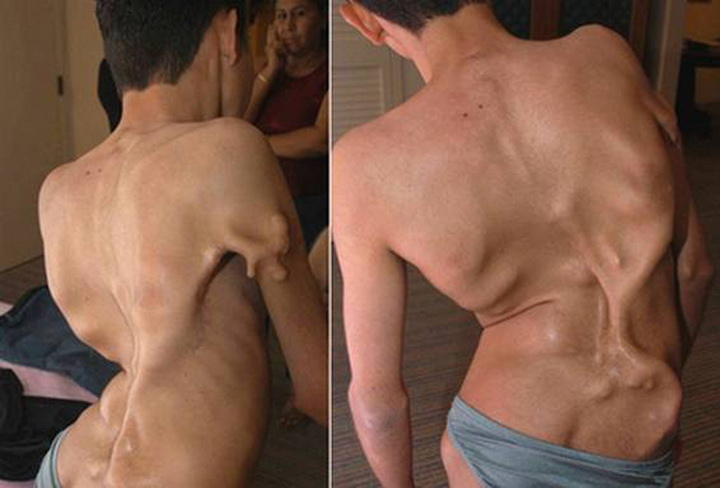
Синдром Марфана — заболевание не такое уж и редкое, как правило, оно проявляется у одного человека из 20 000. Оно представляет собой нарушение в развитии соединительных тканей. Одним из самых распространённых форм отклонения является близорукость, но ещё чаще болезнь проявляется в непропорциональном росте костей в руках и ногах и чрезмерной подвижности коленных и локтевых суставов. Люди с синдромом Марфана, как правило, имеют длинные и тонкие руки и ноги. Реже у больных могут срастаться между собой рёбра, в результате чего грудная клетка или выпирает наружу, или, напротив, западает. Ещё одна проблема — искривление позвоночника